Termodynamika chemiczna to dziedzina na styku chemii i fizyki, badająca efekty energetyczne towarzyszące reakcjom chemicznym, możliwość samorzutnego przebiegu reakcji, warunki przebiegu reakcji odwracalnych (równowaga chemiczna) i podobne zagadnienia.
Zacznijmy od początku. Aby rozpatrywać przebieg reakcji, a w szczególności - towarzyszące jej efekty energetyczne, należy tę reakcję gdzieś "umiejscowić". Reakcja może przebiegać w probówce lub kolbie, zawierającej mieszaninę reagentów, może zachodzić w powietrzu, nad pewnym terenem, w wodzie płynącej w rzece, w żywej komórce itp. Każdy obszar wydzielony w przestrzeni, wraz ze znajdującymi się w nim substancjami nazywany jest układem. Pozostała przestrzeń, na zewnątrz układu, to jego otoczenie.

Stan układu określają tzw. parametry stanu, tj. opisujące go wielkości fizyczne - temperatura, ciśnienie, objętość, ilości (np. stężenia) poszczególnych substancji, czasem również inne. Wielkości, które nie zależą od ilości substancji w układzie, to tzw. parametry intensywne; wielkości zależące od ilości substancji, to parametry ekstensywne. Na przykład temperaturę 20°C może mieć 100ml wody, albo 100l wody. Temperatura jest więc parametrem intensywnym, objętość - ekstensywnym. Przykład powyższy pokazuje również, że parametry stanu są w pewnym sensie niezależne od siebie.

Wielkość fizyczna, która zależy od stanu układu, od wszystkich opisujących go parametrów, to funkcja stanu. Wartość funkcji stanu zależy wyłącznie od aktualnego stanu układu, nie zależy od jego historii. Najważniejszą funkcją stanu jest energia wewnętrzna układu, U, to jest suma wszystkich form energii zawartych w układzie (energia kinetyczna cząsteczek i atomów, energia wiązań chemicznych i elektronów w atomach itd.). Ponieważ, energia wewnętrzna jest funkcją stanu, jej zmiana DU = U2 - U 1, niezależnie od drogi, jaką układ został przekształcony ze stanu "1" do stanu "2".
Układ może być w różnym stopniu związany ze swoim otoczeniem. Jeżeli możliwa jest wymiana materii i energii pomiędzy układem a otoczeniem, układ nazywa się otwartym. Układ, który może wymieniać z otoczeniem energię, ale nie wymienia masy, to układ zamknięty. Wreszcie, układ, który nie wymienia z otoczeniem energii, ani masy, to układ izolowany.
Podstawową własnością energii wewnętrznej jest zasada zachowania energii (Mayer, 1842), zwana też I zasadą termodynamiki: We wszystkich procesach zachodzących w układzie izolowanym energia wewnętrzna nie ulega zmianie,
U = const, czyli: DU = 0
W najczęstszej sytuacji, układ oddaje lub pobiera z otoczenia energię na sposób ciepła, Q, może też wykonywać pracę mechaniczną, W. Jak łatwo wykazać, ponieważ praca mechaniczna, to siła razy przesunięcie (W = FDl), siła, to ciśnienie razy pole powierzchni (F = pS), a pole powierzchni razy przesunięcie, to przyrost objętości (SDl = DV), stąd praca W = pDV. Ponieważ układ, który wykonuje pracę, traci energię, stąd konwencja znaku:
W = - pDV
(Uwaga: taki, uproszczony wzór jest prawdziwy, jeżeli podczas całego procesu p = const. W przeciwnym razie, konieczne jest wykonanie operacji matematycznej, zwanej całkowaniem, a sprowadzającej się do zsumowania iloczynów pDV, dla odpowiedniej liczby małych przedziałów, dla których p można uznać za, w przybliżeniu, stałe.)

Jeżeli układ nie wymienia z otoczeniem innych form energii (światło, energia elektryczna), wówczas zasadę zachowania energii można zapisać wzorem:
DU = Q + W, czyli: DU = Q - pDV
Ponieważ wcześniej założyliśmy, że p = const., możemy zapisać:
DU = Qp - pDV, gdzie Q z indeksem "p" oznacza ciepło procesu wymienione w warunkach stałego ciśnienia,
stąd Qp = DU + pDV
W warunkach stałej objętości, niezależnie od stałości lub nie ciśnienia, człon DV = 0, zatem:
QV = DU, gdzie Q z indeksem "V" oznacza ciepło wymienione w warunkach stałej objętości.
W praktyce, istotne zmiany objętości zachodzą podczas reakcji z udziałem reagentów gazowych, i to takich reakcji, w których zmianie ulega liczba moli reagentów w fazie gazowej: pV = nRT (równanie Clapeyrona), stąd, w stałej temperaturze: D(pV) = DnRT, gdzie Dn oznacza zmianę liczby moli reagentów gazowych w czasie reakcji, R - uniwersalna stała gazowa, T - temperatura bezwględna.
Warto zauważyć, że w warunkach izobarycznych (p = const), wyrażenie Qp = DU + pDV, jest równe zmianie wielkości fizycznej: H = U + pV, która, podobnie jak energia wewnętrzna, jest również funkcją stanu układu. Tak zdefiniowana wielkość nosi nazwę entalpii.
Zatem: Qp = DH - ciepło procesu (reakcji) w warunkach izobarycznych jest równe zmianie entalpii - zachowuje się więc podobnie jak funkcje stanu.Ponieważ, z kolei, QV = DU, zatem można sformułować następujące prawo termodynamiki chemicznej:
Prawo Hessa (1840): Ciepło reakcji w warunkach izobarycznych lub izochorycznych nie zależy od drogi, którą biegnie reakcja, a jedynie od stanu poczatkowego i końcowego reakcji.
Ponieważ wyznaczenie bezwzględnej wartości energii wewnętrznej lub entalpii jest bardzo trudne, jeżeli nie wręcz niemożliwe, stosuje się więc standardowe wartości DU i DH, dla których poziomem odniesienia, poziomem "zerowym" jest układ zawierający pierwiastki chemiczne w stanie standardowym, w ich trwałych postaciach. Za warunki standardowe przyjmuje się T = 298 K i, zależnie od autora, p = 101325 Pa lub p = 100000Pa.
Dla każdej substancji można wyznaczyć tzw. standardową entalpię tworzenia, DH°, to jest zmianę entalpii w reakcji tworzenia 1 mola substancji z pierwiastków w warunkach standardowych, np.
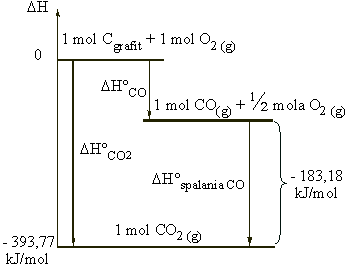
Cgrafit + O2 (g).= CO2 (g), DH°CO2 = - 393,77 kJ/mol
Znając dodatkowo zmianę entapii w warunkach standardowych dla reakcji spalania mola tlenku węgla do dwutlenku, można obliczyć entalpię tworzenia tlenku węgla, trudną do bezpośredniego wyznaczenia doświadczalnego.
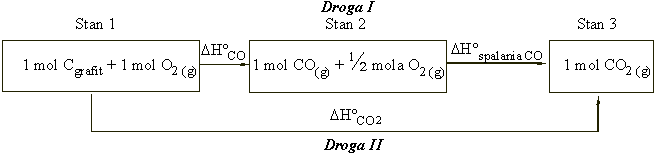
Z prawa Hessa wynika, że całkowita zmiana entalpii przy przejściu ze stanu 1. do stanu 3., jest identyczna, niezależnie od drogi reakcji, zatem:
DH°CO2 = DH°CO + DH°spalania CO , a stąd: DH°CO = DH°CO2 - DH°spalania CO
Podobne rezultaty uzyskamy stosując następujący wzór na zmianę entalpii w reakcji chemicznej:

Inaczej mówiąc - zmiana entalpii w reakcji jest równa sumie entalpii tworzenia produktów (z uwzględnieniem liczby moli każdego z nich) minus suma entalpii tworzenia wszystkich substratów. Np. dla reakcji spalania CO:
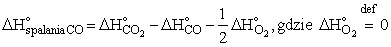
Wiedząc, że entalpia spalania CO wynosi - 183,18 kJ/mol, można obliczyć entalpię tworzenia CO, co proponuję wykonać, jako ćwiczenie.
Inny sposób graficznego przedstawienia zależności energetycznych w opisywanym ciągu reakcji przedstawia diagram:
A oto inny przykład zastosowania prawa Hessa - obliczenie standardowej entalpii tworzenia cyklopropanu, na podstawie entalpii spalania tego związku do dwutlenku węgla i ciekłej wody:
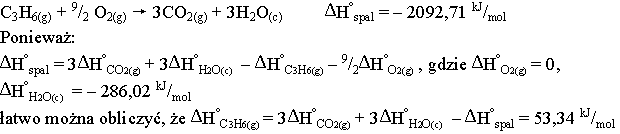
Dodatnia wartość entalpii tworzenia wskazuje, że utworzenie związku wymaga jest procesem endotermicznym, związek jest nietrwały termodynamicznie.
Entalpia rekacji zależy od temperatury. Zależność tę wyraża prawo Kirchhoffa. Pełna wersja tego prawa jest dość skomplikowana, bo uwzględnia zależność tzw. pojemności cieplnej substancji od temperatury. Pojemność cieplna określa ilość ciepła potrzebną do ogrzania określonej ilości substancji o 1 stopień, w określonych warunkach. Na przykład molowa pojemność cieplna pod stałym ciśnieniem, Cp, określa ilość ciepła potrzebną do ogrzania o 1 stopień, w warunkach izobarycznych, 1 mola substancji. Jeżeli rozpatruje się zależność entalpii reakcji od temperatury w niewielkim przedziale temperatur, w którym pojemność cieplna ma stałą wartość, prawo Kirchhoffa można sformułować w postaci uproszczonej:

Przykład: Standardowa entapia tworzenia pary wodnej w temperaturze 298 K wynosi DH°H2O(g) = - 241,82 kJ/mol,
pojemności cieplne reagentów: CpH2O(g) = 33,58 J/(mol·K), CpH2 = 28,84 J/(mol·K), CpO2 = 29,37 J/(mol·K)
Obliczyć DH°H2O(g) w temperaturze 373 K.
Diagram energetyczny dla tego przykładu ma następującą postać:
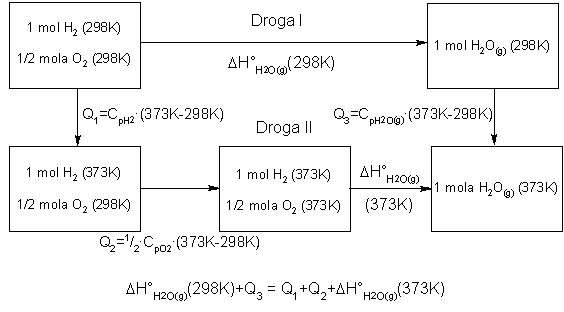
(Odpowiedź: -242,57 kJ/mol)
Na co dzień obserwujemy zwykle zjawiska i procesy, w których energia układu maleje. Efekt cieplny reakcji nie wyjaśnia jednak, dlaczego samorzutnie mogą przebiegać również reakcje endotermiczne, w których energia układu rośnie. Okazuje się, że do opisu przebiegu reakcji (i innych procesów) potrzebna jest jeszcze jedna wielkość fizyczna - funkcja stanu, opisująca aspekt kierunku reakcji. Rozważmy następujący przykład. W układzie znajduje się 1 kg lodu o temperaturze 0°C. Po pewnym czasie ten sam układ zawiera 1 kg ciekłej wody o temperaturze 0°C. W tym czasie układ pochłonął z otoczenia odpowiednią ilość ciepła topnienia lodu, Q. Ponieważ temperatura jest miarą średniej energii kinetycznej cząsteczek, uznać należy, że w układzie całe pochłonięte ciepło zużyte zostało na zmianę struktury mikroskopowej - z uporządkowanego układu krystalicznego, w znacznie mniej uporządkowaną ciecz. W procesie wzrosło "nieuporządkowanie" układu. Funkcja stanu, która opisuje ilościowo "nieuporządkowanie" układu, a ściślej, tzw. prawdopodobieństwo termodynamiczne, nosi nazwę entropii, S.
W każdym samorzutnym procesie entropia rośnie. Jeżeli nawet w obserwowanym układzie rośnie uporządkowanie, dzieje się to kosztem wzrostu entropii otoczenia. W procesie przebiegającym w warunkach odwracalnych (układ w równowadze),
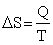 .
. .
.W procesach samorzutnych, jak wspomniano wyżej, entropia wzrasta, w równowadze - entropia osiąga wartość maksymalną. Jest to konsekwencja tzw. II zasady termodynamiki, której jedno ze sformułowań brzmi: W procesach przebiegających samorzutnie, w kierunku osiągnięcia przez układ równowagi, rośnie nieuporządkowanie układu i maleje zdolność układu do wykonania pracy zewnętrznej.
Wyrażając ciepło procesu, jako iloczyn zmiany entropii i temperatury, można zapisać sformułowaną wcześniej I zasadę termodynamiki w następujący sposób:
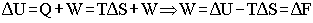 ,
,gdzie F oznacza funkcję stanu, zwaną energią swobodną, F = U - TS. W procesie izotermicznym W = DF, w procesie izobaryczno-izochorycznym, gdy W = 0, DF = 0. Inaczej mówiąć, DF można określić, jako tę część energii wewnętrznej, którą można zamienić na pracę.
Analogicznie definiuje się entalpię swobodną: G = H - TS. W procesach izobaryczno-izotermicznych DG = 0. DG jest wielkością, która określa pracę nieobjętościową wykonaną przez układ, np. pracę elektryczną, co jest szczególnie ważne w elektrochemii.
Podobnie jak entropia, w procesach samorzutnych F i G zawsze maleją (DF < 0, DG < 0), w równowadze osiągają wartości minimalne, a DF = 0, DG = 0
Standardową zmianę entalpii swobodnej w reakcji chemicznej oblicza się analogicznie, jak zmianę entalpii. W tablicach znaleźć można wartości standardowych entalpii i entalpii swobodnych tworzenia najważniejszych związków chemicznych, a także bezwzględne wartości entropii molowych, również pierwiastków chemicznych. Obliczenie entropii jest możliwe, bowiem (zgodnie z tzw. III zasadą termodynamiki) można przyjąć, że wartość zerową osiąga ona dla każdej substancji w stanie krystalicznym, w temperaturze zera bezwzględnego.
Znajomość DG reakcji pozwala na przewidywanie możliwości jej samorzutnego przebiegu. Ponieważ zmiana entalpii i zmiana entropii mogą mieć znaki "+" lub "-", a całe wyrażenie zależy od temperatury, istnieją cztery możliwe przypadki:
| możliwa w dowolnej temperaturze | |||
| niemożliwa w jakiejkolwiek temperaturze | |||
| możliwa w dostatecznie niskiej temperaturze | |||
| możliwa w dostatecznie wysokiej temperaturze |
Przykład rachunkowy.
Zakładając, że zmiany entalpii i entropii w podanej reakcji nie zależą znacząco od temperatury, obliczyć wartość zmiany entalpii swobodnej podanej reakcji w temperaturach; 25, 500 i 1500°C.
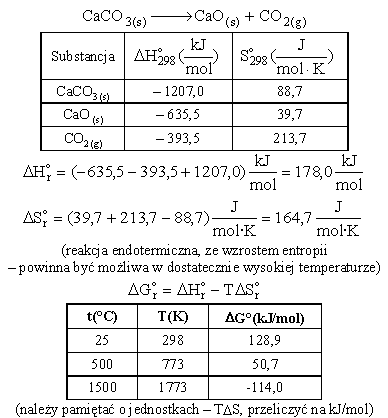
Podane wzory dotyczą substancji czystych, w ilościach wynikających z równania reakcji. Zmiana entalpii swobodnej dotyczy sytuacji, gdy reakcja zachodzi do końca. Jednakże wiele reakcji osiąga stan równowagi chemicznej, a ilości reagentów wynikają ze stałej równowagi. Okazuje się, że istnieją wzory pozwalające obliczyć entalpię swobodną substancji w mieszaninie, w szczególności w roztworze. Wzory te mają postać, w uproszczeniu:
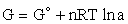 ,
,gdzie G° oznacza entalpię swobodną w warunkach standardowych, n - liczbę moli, R - uniwersalną stałą gazową (R = 8,314J/(mol·K)), natomiast a oznacza tzw. aktywność substancji. Nie wchodząc w niezbyt jasne szczegóły, aktywność należy rozumieć jako miarę ilości substancji w stosunku do ilości standardowej. Dla gazów jest to ciśnienie cząstkowe wyrażone bezwymiarowo, jako wielokrotność ciśnienia standardowego (z uwzględnieniem współczynników aktywności, wyznaczonych eksperymentalnie, a istotnych tylko dla dużych ciśnień); dla roztworów rozcieńczonych aktywność substancji rozpuszczonej jest równa stężeniu molowemu (wyrażonemu bezwymiarowo, z zastrzeżeniem jak wyżej), dla czystych cieczy i ciał stałych aktywność wynosi 1. Symbol "ln" oznacza logarytm naturalny, czyli logarytm o podstawie e = 2,718...- niestety czytelnicy nie znający pojęcia logarytmu powinni w tym miejscu przerzucić się na lekturę podręcznika matematyki).
Dla reakcji w stanie równowagi chemicznej:
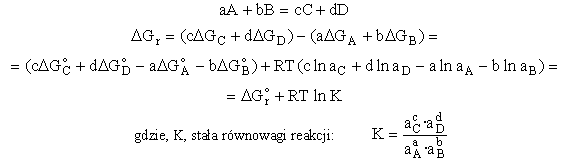
(logarytm iloczynu jest równy sumie logarytmów, logarytm ilorazu - różnicy). Zatem, jeżeli układ jest w stanie równowagi chemicznej, co dopowiada wartości DG = 0, zapisać można:
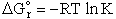 .
.Równanie powyższe pozwala obliczyć zarówno DG° reakcji na podstawie znajomości stałej równowagi, jak i odwrotnie - przewidzieć możliwość odwracalności (lub kierunku) reakcji w określonych warunkach, na podstawie znajomości parametrów termodynamicznych reagentów.